What the Research Actually Says
Epigenetics: Most popular advice about accelerated learning is recycled from a small body of peer-reviewed cognitive science research.
The canonical synthesis is Dunlosky, Rawson, Marsh, Nathan, and Willingham's 2013 paper "Improving Students' Learning With Effective Learning Techniques" in Psychological Science in the Public Interest, which evaluated ten common study techniques against experimental evidence.
Two techniques earned the "high utility" rating: practice testing (active recall) and distributed practice (spaced repetition). Three earned moderate utility. Five earned low utility.
The techniques in the low-utility category include almost everything learners naturally gravitate toward: highlighting, rereading, summarizing, imagery for text, and the keyword mnemonic. The gap between what feels productive and what produces durable learning is the central insight of the last four decades of learning science.
"The most effective learning techniques are not the ones that feel easiest. They are the ones that generate desirable difficulties during acquisition and pay dividends at retrieval."
- Robert Bjork, UCLA Learning and Forgetting Lab, Making Things Hard on Yourself, But in a Good Way (2011)
This article covers nine techniques. Each is grounded in peer-reviewed research, each has documented time benchmarks, and each can be implemented without specialized tools.
The Nine Techniques in One Table
| Technique | Research Utility | Time Investment | Best For |
|---|---|---|---|
| Active Recall | High | Low | Factual knowledge, concepts |
| Spaced Repetition | High | Low (long-term) | Large fact stores, language |
| Feynman Technique | Moderate-High | Moderate | Conceptual understanding |
| Deliberate Practice | High | High | Skill acquisition |
| Interleaving | Moderate | Low | Discrimination problems |
| Dual Coding | Moderate | Low | Complex systems, diagrams |
| Chunking | Moderate-High | Moderate | Sequences, procedures |
| 80/20 Curation | N/A (strategic) | Low | Breadth before depth |
| Deep Work Blocks | High (correlational) | High | All complex learning |
1. Active Recall (Retrieval Practice)
Active recall is the act of producing information from memory rather than reviewing it. The gap between "can I recognize this" and "can I produce this" is where durable learning forms.
Karpicke and Roediger's 2008 paper in Science, "The Critical Importance of Retrieval for Learning," showed that one round of self-testing produced retention 50 percent higher after one week than four rounds of rereading.
How to apply:
- After reading, close the source and write what you remember.
- Use flashcards, but generate the cards yourself rather than using premade sets.
- Practice essays, problem sets, and simulations rather than re-reading solutions.
Time benchmark: A 25-minute active-recall session produces better retention than a 60-minute rereading session. The differential compounds over weeks.
2. Spaced Repetition
Ebbinghaus's forgetting curve, published in 1885, established that memory decays exponentially without retrieval. Spaced repetition schedules retrievals at increasing intervals (1 day, 3 days, 7 days, 14 days, 30 days) matched to the decay rate.
Cepeda, Pashler, Vul, Wixted, and Rohrer's 2006 meta-analysis in Psychological Bulletin of 254 studies found that spaced practice outperformed massed practice by an average effect size of 0.42 standard deviations.
Tools:
- Anki (free, SRS algorithm, desktop and mobile)
- Quizlet (easier onboarding, less customizable)
- Physical Leitner box system (zero-tech, three to five boxes at increasing review intervals)
Time benchmark: For a typical professional certification with 500 to 1,000 discrete facts, 15 to 20 minutes daily of Anki review for 90 to 180 days produces near-ceiling retention.
3. Feynman Technique
Richard Feynman's reported study method: pick a concept, explain it in plain language as if teaching a twelve-year-old, identify gaps where the explanation breaks down, return to source material on those gaps, and repeat until the explanation flows without technical jargon.
The technique is a hybrid of active recall and self-explanation, the latter well supported by Chi, de Leeuw, Chiu, and LaVancher's 1994 paper in Cognitive Science, which found that learners who self-explained problem-solving steps solved transfer problems at roughly twice the rate of learners who simply read worked examples.
How to apply:
- Write the concept name at the top of a blank page.
- Explain it as if to a bright 12-year-old, in plain language, with analogies.
- Identify sentences where you slipped into jargon or hand-waved.
- Return to the source material and fill the gaps.
- Rewrite until the explanation flows.
Time benchmark: 30 to 60 minutes per concept. Slower per concept than passive review, but retention and transfer are dramatically higher.
4. Deliberate Practice
Anders Ericsson's research on expert performance, synthesized in Peak: Secrets from the New Science of Expertise (2016), distinguishes deliberate practice from mere experience. Deliberate practice requires: a specific sub-skill goal, immediate feedback, operation at the edge of current capability, and full attention.
Ericsson's studies of violinists at the Berlin Music Academy found that by age 20, the top performers had accumulated roughly 10,000 hours of deliberate practice, while the good-but-not-elite performers had accumulated 8,000 hours, and the teacher-track group had 4,000.
The 10,000-hour figure popularized by Malcolm Gladwell is an average, not a threshold, and applies to domains where expert performance is well-defined.
Key distinction: 10 hours of deliberate practice (focused, effortful, feedback-rich) produces more skill gain than 100 hours of casual practice.
5. Interleaving
Instead of practicing one skill or topic until mastered, interleaving mixes related-but-distinct problems in the same session.
Rohrer, Dedrick, and Stershic's 2015 study in the Journal of Educational Psychology on middle school math found that students practicing interleaved problem types outperformed students practicing in blocks by 25 percentage points on a delayed test.
The mechanism: interleaving forces the learner to discriminate between problem types and select the right solution method, a cognitive operation that blocked practice skips.
Application example: When studying for a certification exam, mix question types within each study session rather than completing all of one section before moving to the next.
6. Dual Coding
Paivio's dual coding theory, introduced in 1971 and validated in hundreds of subsequent studies, holds that information encoded in both verbal and visual modalities is remembered better than information encoded in one.
Richard Mayer's multimedia learning research at UC Santa Barbara, documented across 20 years of experiments, shows consistent effect sizes of d = 0.5 to 1.0 for dual-coded presentations.
Application: When studying a complex system, draw it. Concept maps, flowcharts, and hand-drawn diagrams outperform pure text study for systems-level content. The drawing does not need to be artistic. Rough shapes and arrows are sufficient.
7. Chunking
George Miller's 1956 paper "The Magical Number Seven, Plus or Minus Two" established that working memory holds roughly seven units. Chunking is the process of building larger units from smaller ones so that working memory can hold more functional content.
Chess research provides the clearest example. Chase and Simon's 1973 study in Cognitive Psychology found that chess grandmasters could recall 20 to 25 pieces from a mid-game position after a five-second look, while novices recalled 5 to 6.
When the pieces were placed randomly (destroying the chunks), grandmasters performed no better than novices.
Application: Break new material into 5 to 9 meaningful units, learn each unit, then combine. For a programming language, learn syntax (chunk 1), control flow (chunk 2), data structures (chunk 3), and so on. For a medical topic, organize by system, then condition, then presentation.
8. 80/20 Curation (Strategic, Not Cognitive)
The Pareto principle applied to learning: in most domains, 20 percent of the material produces 80 percent of the functional capability. The technique predates modern cognitive science but is complementary to it.
Tim Ferriss's The 4-Hour Chef popularized the method for adult learning: before studying a domain, ask which 20 percent of the content is most frequently used, most often tested, or most load-bearing.
For language learning, the highest-frequency 1,000 words cover roughly 80 percent of typical conversation. For cybersecurity certifications, core concepts appear in 60 to 70 percent of exam questions.
9. Deep Work Blocks
Cal Newport's Deep Work (2016) synthesizes research on attention and productivity into the argument that uninterrupted, focused work is both rarer and more valuable than most knowledge workers recognize.
The research base includes Sophie Leroy's 2009 work on attention residue and Gloria Mark's 2008 research at UC Irvine on task-switching costs, which found that interrupted workers took an average of 23 minutes and 15 seconds to return to the original task.
Practical benchmark: A 90-minute uninterrupted deep work block produces more learning progress than three 30-minute blocks punctuated by notifications. The research on task-switching costs is robust across decades and methodologies.
Tools that remove friction from deep work include distraction blockers (Freedom, Cold Turkey), physical environment design (phone in another room), and markdown-based note systems that do not require context-switching into cloud apps.
Four Study Schedules
The schedules below are calibrated for typical adult learners with 10 to 25 hours per week to invest.
Certification Study (10 weeks, 15 hours per week)
- Week 1 to 2: 80/20 curation. Map the exam objectives. Identify high-weight domains.
- Week 3 to 6: Active recall and spaced repetition. Build Anki deck of core facts. Study 60 to 90 minutes daily.
- Week 7 to 8: Interleaved practice testing. Mixed-topic practice exams.
- Week 9: Feynman technique on the five weakest areas.
- Week 10: Rest, light review, and exam.
Language Acquisition (6 months to conversational fluency)
- Month 1: 1,000 highest-frequency words via spaced repetition (Anki). 30 minutes daily.
- Month 2 to 3: Comprehensible input (podcasts, children's shows) 30 minutes daily, plus continued SRS.
- Month 4 to 5: Speaking practice with tutors (iTalki, Preply), 60 to 90 minutes weekly.
- Month 6: Immersion blocks (movies, books, conversation partners).
Programming Skill (3 to 6 months to employable)
- Month 1: Core syntax and data structures via CS50x or equivalent. Problem sets daily.
- Month 2: Interleaved projects. Rotate between frontend, backend, and data projects.
- Month 3 to 4: One shipped project per month, deployed publicly.
- Month 5 to 6: Open-source contributions and interview preparation with active recall on system design.
Academic Exam (4 weeks intensive)
- Week 1: 80/20 syllabus review, active recall during initial reading.
- Week 2: Spaced repetition on core terminology. Interleaved practice problems.
- Week 3: Feynman technique on the hardest concepts. Deep work blocks of 90 minutes.
- Week 4: Full practice exams under timed conditions. Light review on weak areas.
Benchmarks and Realistic Expectations
| Skill | Realistic Time to Entry Competency | Time to Advanced |
|---|---|---|
| Programming (full-stack) | 800 to 1,500 hours | 3,000 to 5,000 hours |
| Conversational language | 500 to 750 hours (category I) | 2,200+ hours (category IV) |
| Chess (from 0 to 1500 ELO) | 500 to 1,000 hours | 3,000+ hours |
| Musical instrument (basic proficiency) | 600 to 900 hours | 4,000+ hours |
| Data analyst portfolio | 400 to 600 hours | 1,500+ hours |
Language category ratings come from the U.S. Foreign Service Institute, which classifies languages from Category I (Spanish, French, Italian, 600 to 750 hours for a native English speaker) to Category IV (Mandarin, Arabic, Japanese, Korean, 2,200 hours).
Working memory, processing speed, and pattern recognition each predict learning speed in different domains, and self-awareness about which is relatively stronger or weaker shapes study design."The single most important question a serious learner can ask is not 'what should I study' but 'what should I practice retrieving.' Everything else is preparation for the retrieval."
- Peter Brown, Henry Roediger, and Mark McDaniel, Make It Stick: The Science of Successful Learning, 2014
What Does Not Work
The Dunlosky et al. 2013 review was blunt about five widely-used techniques with weak empirical support:
| Technique | Utility Rating | Why It Fails |
|---|---|---|
| Highlighting and underlining | Low | Passive, no retrieval |
| Rereading | Low | Illusion of mastery, no retrieval |
| Summarization | Low (for most learners) | Requires skill most students lack |
| Keyword mnemonic | Low | Narrow applicability, fragile |
| Imagery for text | Low | Limited to concrete text |
The common feature of low-utility techniques is that they feel productive while requiring no retrieval. The common feature of high-utility techniques is that they require retrieval under some difficulty.
Frequently Asked Questions
Is the Feynman technique really worth the extra time per concept?
Yes, for any material where conceptual understanding matters more than memorization of isolated facts. The underlying mechanism is self-explanation, which Chi and colleagues demonstrated in a 1994 Cognitive Science paper produces roughly double the transfer-problem success rate compared to worked-example study alone.
The technique is slower per concept than passive review. The retention and transfer advantage compounds. For pure memorization tasks such as vocabulary or medical anatomy terminology, spaced repetition outperforms the Feynman technique.
For conceptual domains such as programming, statistics, economics, physics, and machine learning, the Feynman approach is superior. The combination of both, Feynman for understanding and spaced repetition for retention, is the most robust pattern documented in the cognitive science literature.
How much faster can someone realistically learn using these techniques?
The cleanest experimental comparisons, from Karpicke and Roediger's 2008 Science paper on retrieval practice, show roughly 50 percent better retention from active recall versus rereading across comparable time investment.
Cepeda and colleagues' 2006 meta-analysis on spaced versus massed practice found an effect size of 0.42 standard deviations.
Stacking active recall, spaced repetition, interleaving, and deep work blocks produces compounding gains that, across a 10-week study program, can plausibly produce 2x to 3x the retained learning compared to an equivalent time investment in highlighting and rereading.
The ceiling is not infinite. Individual differences in working memory and processing speed set a hard limit on rate of skill acquisition, and most dramatic claims of accelerated learning (learn Mandarin in 3 months, become a programmer in 30 days) ignore the task-hour research showing realistic time ranges.
What is the single most impactful change for someone starting from a highlighter-and-reread background?
Replace one hour of rereading per week with one hour of self-testing. The switch is simple, costs no additional time, and produces measurable retention gains within the first study cycle.
Practical implementation: after reading a textbook chapter or watching a lecture, close the source, set a 20-minute timer, and write everything you can recall about the material.
Then open the source and fill the gaps. This single practice, repeated across a study program, is the highest-leverage change documented in the learning science literature.
It replicates across elementary school, medical school, professional certification, and adult learning contexts. The resistance most learners feel to self-testing (because it feels harder and exposes what they do not know) is precisely what makes it effective.
Bjork's desirable-difficulty principle predicts that productive struggle in acquisition predicts durable retrieval.
Sources & Further Reading
Dunlosky, J., Rawson, K. A., Marsh, E. J., Nathan, M. J., & Willingham, D. T. (2013). Improving students' learning with effective learning techniques: Promising directions from cognitive and educational psychology. Psychological Science in the Public Interest, 14(1), 4-58. DOI: 10.1177/1529100612453266
Karpicke, J. D., & Roediger, H. L. (2008). The critical importance of retrieval for learning. Science, 319(5865), 966-968. DOI: 10.1126/science.1152408
Cepeda, N. J., Pashler, H., Vul, E., Wixted, J. T., & Rohrer, D. (2006). Distributed practice in verbal recall tasks: A review and quantitative synthesis. Psychological Bulletin, 132(3), 354-380. DOI: 10.1037/0033-2909.132.3.354
Ericsson, K. A., Krampe, R. T., & Tesch-Romer, C. (1993). The role of deliberate practice in the acquisition of expert performance. Psychological Review, 100(3), 363-406. DOI: 10.1037/0033-295X.100.3.363
Rohrer, D., Dedrick, R. F., & Stershic, S. (2015). Interleaved practice improves mathematics learning. Journal of Educational Psychology, 107(3), 900-908. DOI: 10.1037/edu0000001
Chi, M. T. H., de Leeuw, N., Chiu, M. H., & LaVancher, C. (1994). Eliciting self-explanations improves understanding. Cognitive Science, 18(3), 439-477. DOI: 10.1207/s15516709cog1803_3
Mayer, R. E. (2009). Multimedia Learning (2nd ed.). Cambridge University Press. DOI: 10.1017/CBO9780511811678
Bjork, R. A., Dunlosky, J., & Kornell, N. (2013). Self-regulated learning: Beliefs, techniques, and illusions. Annual Review of Psychology, 64, 417-444. DOI: 10.1146/annurev-psych-113011-143823
Frequently Asked Questions
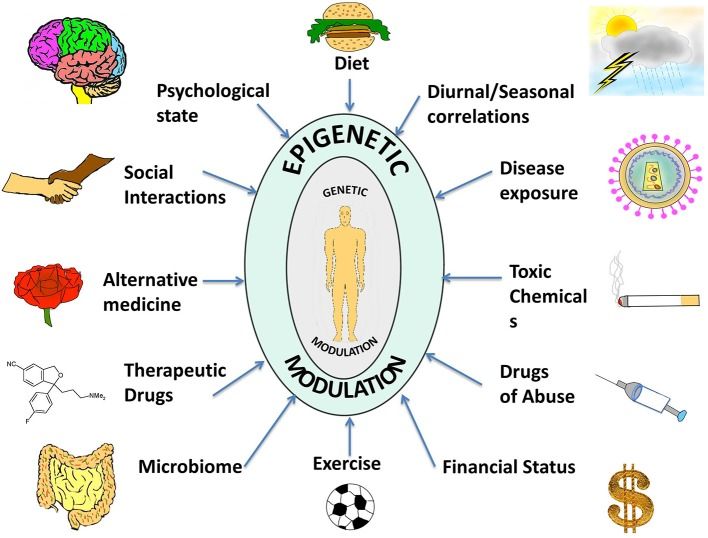
What is epigenetics and how is it different from genetics?
Genetics is the study of DNA sequences, the order of the four chemical bases (adenine, thymine, cytosine, guanine) that encode the instructions for building proteins. Every cell in your body contains the same DNA sequence: the same genetic instructions that were present in the fertilized egg you developed from. Epigenetics is the study of the system that determines which parts of those instructions are actually read in any given cell, at any given time, in response to any given conditions, without changing the underlying DNA sequence. The word ‘epigenetics’ literally means ‘above genetics,’ reflecting that epigenetic regulation operates at a level above the DNA sequence itself. Conrad Waddington, who coined the term in 1942, used it in a broader developmental biology sense: the study of how genes interact with the environment to produce the phenotype. In contemporary molecular biology, epigenetics refers specifically to heritable changes in gene expression that do not involve changes to the DNA sequence. The key word is ‘heritable’: epigenetic marks can be passed from a cell to its daughter cells through cell division, maintaining patterns of gene expression across many rounds of replication. The practical significance of this is profound: it explains how a liver cell and a neuron, despite containing identical DNA, are radically different in structure and function. They have different epigenetic marks that determine which genes are expressed in each cell type. These marks were established during development and are stably maintained throughout the organism’s life. Epigenetics is also the mechanism by which environmental experiences, stress, nutrition, toxin exposure, can alter gene expression patterns, sometimes in long-lasting ways.
How do DNA methylation and histone modification control gene expression?
DNA methylation is the addition of a methyl group (CH3) to the cytosine base in DNA, almost always at specific sites called CpG dinucleotides (where cytosine is followed by guanine in the DNA sequence). Approximately 80 percent of CpG sites in the human genome are methylated under normal conditions. Promoter regions, the DNA sequences that control whether a gene is transcribed into RNA, are often associated with clusters of CpG sites called ‘CpG islands.’ When these islands are methylated, the gene is generally silenced: the methylation physically and biochemically blocks the transcriptional machinery from reading the gene, and also recruits proteins that further compact the DNA into a structure that is inaccessible to transcription factors. Conversely, unmethylated promoter CpG islands are associated with transcriptionally active genes. DNA methylation is relatively stable, it persists through cell division, and can be detected using a technique called bisulfite sequencing. Histone modification is a complementary layer of epigenetic regulation. DNA in the cell nucleus is not free; it is wrapped around proteins called histones, forming a compact structure called chromatin. The tail regions of histones, segments that protrude from the nucleosome, are subject to many types of chemical modification: acetylation, methylation, phosphorylation, ubiquitination, and others. Histone acetylation (the addition of acetyl groups) generally loosens the chromatin structure, making DNA more accessible to transcriptional machinery and therefore increasing gene expression. Histone methylation has variable effects depending on which amino acid is methylated and whether one, two, or three methyl groups are added. The combinatorial ‘histone code’ created by the pattern of modifications across many sites is read by regulatory proteins that either activate or repress transcription. Together, DNA methylation and histone modifications work as coordinated, mutually reinforcing layers of gene regulation.
Can stress and trauma actually change your epigenome?
Yes, and this is one of the most active and consequential areas of current epigenetics research. The clearest evidence comes from studies in model organisms, where experimental conditions can be controlled in ways impossible in humans. Michael Meaney and Moshe Szyf’s seminal research at McGill University, published in a landmark 2004 Nature Neuroscience paper with Ian Weaver and colleagues, showed that the quality of maternal care in rats, specifically, how much the mother licked and groomed her pups, produced lasting differences in the epigenetic regulation of the glucocorticoid receptor gene in the hippocampus of offspring. High-licking mothers produced offspring with lower methylation of the glucocorticoid receptor promoter, more receptor expression, and lower stress reactivity throughout life. Low-licking mothers produced the opposite pattern. Crucially, this epigenetic difference was not fixed at birth, it was established in early postnatal life through experience, and it could be pharmacologically reversed in adult animals using a histone deacetylase inhibitor. In humans, the evidence is more complex and the ethical constraints on experimental study are obviously prohibitive. Rachel Yehuda’s research on Holocaust survivors and their children found altered cortisol levels and methylation patterns on stress-related genes (particularly FKBP5, which regulates glucocorticoid receptor sensitivity) in both survivors and their offspring, suggesting that the biological signature of extreme trauma may transmit to the next generation. These findings have attracted significant attention but also scrutiny: sample sizes are small, replication in independent cohorts is limited, and the causal mechanisms are not fully established. Other research has shown epigenetic effects of early childhood adversity, socioeconomic deprivation, and chronic psychological stress on methylation patterns at relevant gene loci. The evidence collectively supports the conclusion that stress and trauma can alter gene regulation in lasting ways, though the full extent and mechanisms of these effects remain under investigation.
What is the epigenetic clock and how does it measure biological aging?
Steve Horvath’s 2013 Genome Biology paper introduced the concept of the ‘epigenetic clock’: a mathematical model that uses DNA methylation patterns at specific CpG sites to predict chronological age with remarkable accuracy. Horvath analyzed methylation data from over 8,000 samples representing 51 different tissue types and cell types, and identified a set of 353 CpG sites whose methylation levels changed in a consistent, predictable pattern across the human lifespan. The model using these sites could predict chronological age from a DNA sample with a median absolute error of approximately 3.6 years, better than any other biological marker known at the time. Subsequent research developed additional epigenetic clocks, the Hannum clock, the PhenoAge clock, GrimAge, that not only predict chronological age but also independently predict mortality risk, disease onset, and other health outcomes. These second-generation clocks measure what has been called ‘biological age’ or ‘epigenetic age’: a measure of how rapidly the epigenome is aging, which can diverge from chronological age. Individuals whose epigenetic age is older than their chronological age (positive ‘epigenetic age acceleration’) show higher risks of a range of age-related diseases and earlier mortality, even after controlling for chronological age and other known risk factors. The epigenetic clock accelerates in response to chronic stress, adversity, trauma, smoking, and obesity, and appears to decelerate in response to exercise, healthy diet, and other health-promoting behaviors. The biological significance of the clock, whether it is a driver of aging or merely a marker of underlying processes, is still being determined. But its predictive validity for health outcomes makes it one of the most powerful tools yet developed for measuring biological aging.
Can epigenetic changes be inherited by your children?
This is the most contested question in contemporary epigenetics, and the answer depends critically on the organism, the type of epigenetic mark, and the degree of evidence required. In plants, transgenerational epigenetic inheritance, the transmission of epigenetic marks from parents to offspring, is well established and plays an important role in adaptation. In animals, including mammals, the situation is more complicated because of a process called epigenetic reprogramming: when germ cells (sperm and eggs) form and when the embryo develops after fertilization, most epigenetic marks are systematically erased and reset. This reprogramming is the main reason that most environmentally acquired epigenetic changes do not pass to offspring. However, there are documented exceptions. Some specific genomic regions, particularly imprinted genes and certain retrotransposons, escape reprogramming and can transmit epigenetic marks across generations. Lars Olov Bygren’s work on the Overkalix cohort in Sweden showed associations between paternal grandparents’ food availability in a specific developmental window and descendants’ mortality risk, a provocative finding suggesting that nutritional experiences might transmit across generations through some biological mechanism, though whether epigenetics is the actual channel is debated. Brian Dias and Kerry Bhattacharya’s 2014 Nature Neuroscience paper showed that mice conditioned to fear a specific odor transmitted enhanced sensitivity to that odor to their offspring and grandoffspring, apparently through methylation changes at the relevant olfactory receptor gene in sperm. This finding attracted enormous attention but has not been fully replicated in all its claimed details. Rachel Yehuda’s Holocaust research suggests inter-generational transmission in humans, but the sample sizes and methodological complexity make firm conclusions difficult. The current scientific consensus is that while some specific transgenerational epigenetic transmission occurs in mammals, it is not the general rule, and the evidence for robust human transgenerational epigenetic inheritance of acquired characteristics remains limited.
Is epigenetics the same as Lamarckian inheritance?
No, though the confusion is understandable and the distinction matters for both scientific accuracy and avoiding overclaiming. Jean-Baptiste Lamarck’s early nineteenth-century evolutionary theory proposed that acquired characteristics, traits an organism develops in response to its environment during its lifetime, can be directly inherited by offspring. The giraffe’s long neck in Lamarck’s theory elongated through use during the animal’s lifetime, and this acquired elongation was then passed to offspring. This theory was displaced by Darwinian natural selection, which holds that heritable variation arises from random mutation rather than from environmentally directed modification, and that selection acts on this pre-existing heritable variation. The reason epigenetics is not Lamarckian inheritance, in the full sense, is epigenetic reprogramming. When germ cells form in mammals, most epigenetic marks accumulated during the parent’s lifetime are systematically erased. A parent who develops a specific methylation pattern in liver cells in response to a high-fat diet does not generally transmit those liver-cell methylation patterns to offspring. The gametic epigenome is substantially reset with each generation. The exceptions to this rule, cases where some epigenetic marks escape reprogramming and appear in offspring, do have a superficial resemblance to Lamarckian inheritance in that they involve environmentally influenced changes that persist across generations. But they differ in important ways: they affect a small fraction of the epigenome, not all acquired characteristics; the mechanism is not a general inheritance of experience but the escape of specific marks from reprogramming; and the evidence for robust, lasting transmission across multiple generations in mammals is limited. Responsible epigenetics communication distinguishes between the scientifically well-supported finding (environmental experiences can alter gene expression in lasting, sometimes intergenerational ways) and the overclaimed version (acquired characteristics are generally inherited, Lamarckism vindicated).
How is epigenetics involved in cancer?
Epigenetic dysregulation is a universal feature of cancer and is now recognized as a driver of malignancy alongside genetic mutation, not merely a consequence of it. Cancer genomes show two characteristic epigenetic abnormalities that operate simultaneously. The first is global DNA hypomethylation: a widespread, genome-wide reduction in methylation compared to normal tissues. This hypomethylation occurs particularly in repetitive elements and gene bodies, and has several consequences including genomic instability (hypomethylated repetitive elements can move within the genome, causing mutations) and aberrant activation of genes that should be silenced. The second is focal promoter hypermethylation: specific, targeted increases in methylation at the promoter regions of tumor suppressor genes, the genes that normally constrain cell proliferation and induce apoptosis (programmed cell death). Hypermethylation silences these genes, which provides a selective growth advantage to cancer cells analogous to a conventional tumor-suppressing mutation but without changing the DNA sequence. This means that cancer cells can effectively ‘turn off’ tumor suppressor genes through epigenetic silencing rather than through mutation, and the resulting silencing can be heritable through cell division. The reversibility of epigenetic changes, unlike genetic mutations, they do not permanently alter the DNA sequence, has made the epigenome an attractive therapeutic target. Two classes of epigenetic drugs have been approved for clinical use. DNA methyltransferase inhibitors (azacitidine, decitabine) prevent methylation, reactivating silenced tumor suppressor genes and disrupting cancer cell epigenetic patterns. Histone deacetylase inhibitors (vorinostat, romidepsin) alter chromatin structure in ways that promote cancer cell death. Both drug classes are currently approved primarily for hematological malignancies, and research is ongoing into their application in solid tumors and in combination with other therapies.